Pseudomonas aeruginosa is a leading cause of healthcare-associated infections and hospital outbreaks, particularly in high-risk units like ICUs. Traditional typing methods, such as pulsed-field gel electrophoresis, are being replaced by whole-genome sequencing (WGS) as the gold standard for outbreak investigations. WGS provides high-resolution insights through core-genome MLST (cgMLST) and single-nucleotide polymorphism (SNP) analyses, but interpreting genetic relatedness requires careful consideration of epidemiological context.
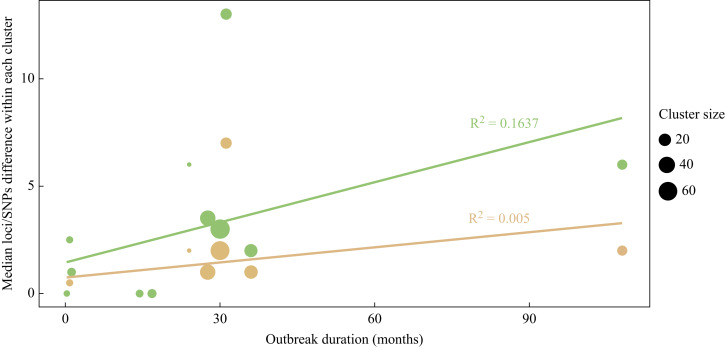
This study aimed to define genomic thresholds for identifying P. aeruginosa transmission chains in hospital outbreaks. By analyzing 14 outbreaks from the University Hospital of Lausanne and reviewing published literature, we found that genetic thresholds alone are insufficient to determine clonality. Factors such as genotype, transmission pathways, environmental reservoirs, and hypermutators significantly influence genetic diversity within outbreaks. For example, environmental reservoirs and hypermutators can lead to higher genetic variation, even among clonally related isolates.
While WGS offers unparalleled discriminatory power, its results must be integrated with epidemiological data to accurately assess transmission dynamics. Standardizing methods, such as using the oldest outbreak isolate as a reference for SNP calling, could improve consistency. As WGS becomes more accessible, it may emerge as the standard for P. aeruginosa outbreak investigations, providing deeper insights into transmission and evolution.
For more information, see the published paper.